Lysophosphatidic acid receptor 1 (LPA1) in pulmonary fibrosis fact sheet

While currently approved therapies can slow the decline in lung function caused by pulmonary fibrosis (PF), unmet needs remain. Fueled by our passion to help more patients, we are driven to tailor treatments to individual needs and help relieve the burdens of pulmonary fibrosis.
Understanding pulmonary fibrosis
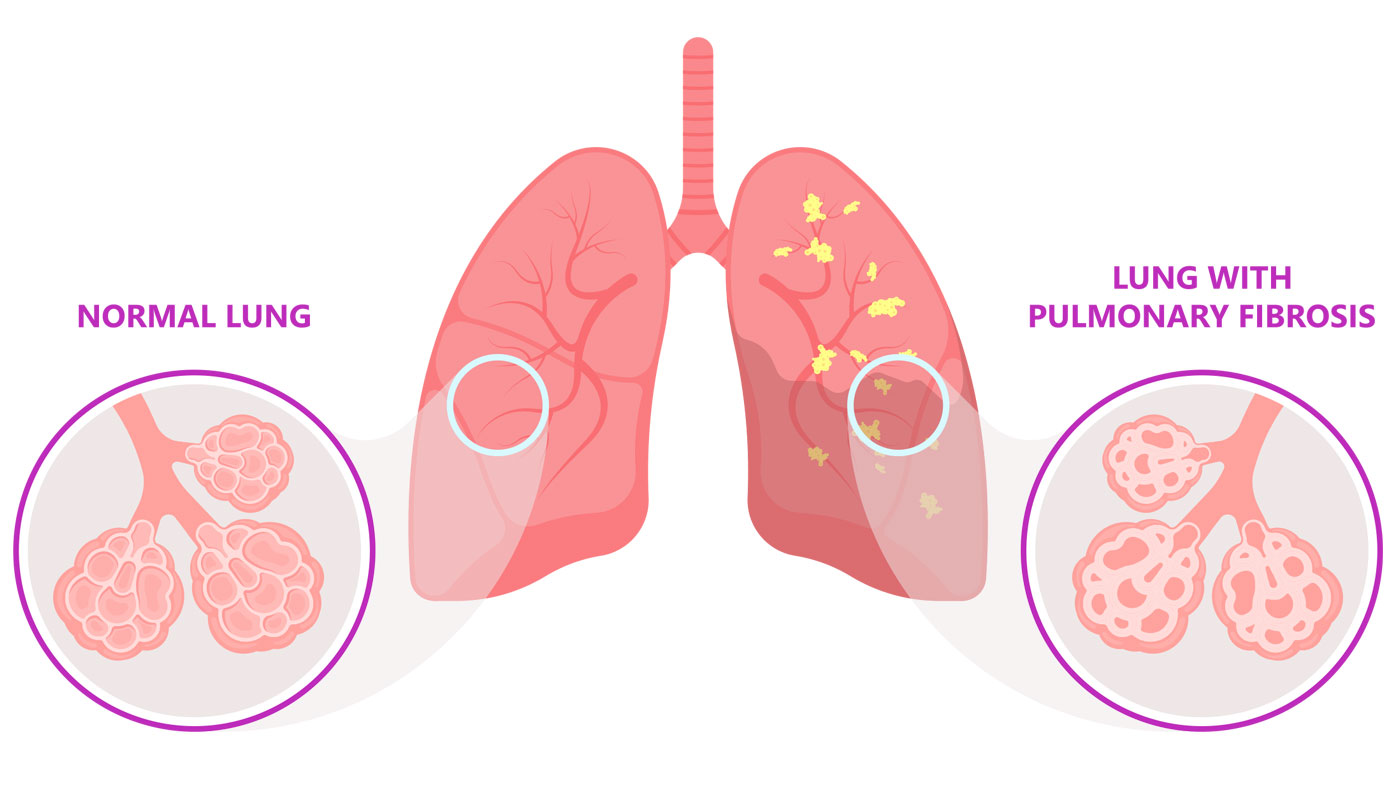
Pulmonary fibrosis (fibrosis occurring in the lungs) is a chronic, life-threatening interstitial lung disease (ILD) that is associated with worsening respiratory symptoms and reduced quality of life.1,2
Fibrosis is defined by the overgrowth, hardening and/or scarring of various tissues.1 It can occur in virtually any organ and results from a maladaptive wound healing response to persistent injury.1,3,4,5
Pulmonary fibrosis occurs when lung tissue becomes damaged and scarred, reducing the lung's ability to expand, collapse and, ultimately, function effectively.6-8 The disease can lead to respiratory failure and death.1,2
Understanding PF subtypes: PPF and IPF
Idiopathic pulmonary fibrosis (IPF) is the most common type of progressive fibrosing ILD.1 Because the cause of IPF is unknown, early diagnosis is challenging, and disease progression is unpredictable, making it difficult to treat. IPF is a fatal disease with a median survival time of 3 to 5 years following diagnosis and a 5-year survival rate of approximately 45%.

Progressive pulmonary fibrosis (PPF) is the clinical designation to describe patients who have ILD with a progressive fibrotic phenotype.9
Many people living with PPF and IPF are physically impaired, experience a progressive decline in lung function, have diffculty performing simple daily activities due to breathlessness and chronic cough and require continuous supplemental oxygen to ease the burden of normal breathing.
Innovation in treatment has been limited with few new therapies approved in more than 10 years.
As a result, a significant need exists for new, transformational therapies.10
LPA in pulmonary fibrosis
Lysophosphatidic acid (LPA) mediates a multicellular response to wound healing and collagen deposition.

Increased LPA levels and activation of LPA1 have been implicated in the pathogenesis of pulmonary fibrosis.11
Preclinical in vitro and in vivo studies found that targeting LPA1 may have an impact in treating lung injury and fibrosis.7,12,13
At Bristol Myers Squibb, we are leveraging our deep expertise in drug discovery and disease biology
to research potential treatments for pulmonary fibrosis and other pulmonology diseases with
high unmet needs.
REFERENCES:
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199-210. doi:10.1002/path.2277
- Selman M, Pardo A. From pulmonary fibrosis to progressive pulmonary fibrosis: A lethal pathobiological jump. Am J Physiol Lung Cell Mol Physiol. 2021;321(3):L600-l607. doi:10.1152/ajplung.00310.2021
- Rockey DC, Bell PD, Hill JA. Fibrosis—a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138-1149. doi:10.1056/NEJMra1300575
- Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020 Nov;587(7835):555-566. doi: 10.1038/s41586-020-2938-9.
- Koudstaal T, Funke-Chambour M, Kreuter M, et al. Pulmonary fibrosis: from pathogenesis to clinical decision-making. Trends Mol Med. 2023 Dec;29(12):1076-1087. doi: 10.1016/j.molmed.2023.08.010.
- Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190(8):867-878. doi:10.1164/rccm.2014030509PP. doi:10.1164/rccm.201403-0509PP
- Swaney JS, Chapman C, Correa LD, et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br J Pharmacol. 2010;160(7):1699-1713.doi:10.1111/j.1476-5381.2010.00828.x
- Ito JT, Lourenço JD, Righetti RF, et al. Extracellular matrix component remodeling in respiratory diseases: What has been found in clinical and experimental studies? Cells. 2019;8(4). doi:10.3390/cells8040342.
- Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: An o cial ATS/ERS/JRS/Alat clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9). doi:10.1164/rccm.202202-0399st
- Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. 2019;20(1):205. doi:10.1186/s12931-019-1161-4
- Smith RA, Kim J, Donnelly DJ, et al. BMS-986327 as a novel PET imaging agent for assessment of LPA1 receptors in IPF. Poster presented at: European Respiratory Society (ERS) International Congress; September 28-October 2, 2019; Madrid, Spain.
- Decato BE, Leeming DJ, Sand JMB, et al. LPA1 antagonist BMS-986020 changes collagen dynamics and exerts antifibrotic effects in vitro and in patients with idiopathic pulmonary fibrosis. Respir Res. 2022 Mar 18;23(1):61. doi: 10.1186/s12931-022-01980-4.
- Cheng PTW, Kaltenbach RF 3rd, Zhang H, et al. Discovery of an Oxycyclohexyl Acid Lysophosphatidic Acid Receptor 1 (LPA1) Antagonist BMS-986278 for the Treatment of Pulmonary Fibrotic Diseases. J Med Chem. 2021 Nov 11;64(21):15549-15581. doi: 10.1021/acs.jmedchem.1c01256.